
Professor Rachel Simmonds FRSB
About
Biography
Biography
Research in my group focusses on the mechanism of action of mycolactone, the lipid-like exotoxin of the Buruli ulcer infectious agent, Mycobacterium ulcerans. Mycolactone has many unusual and fascinating biological functions, and in 2014 we identified its mechanism of action - as an inhibitor of protein translocation into the endoplasmic reticulum. We are now applying this knowledge to understand more about Buruli ulcer, a neglected tropical disease, and basic cell biology. In particular, I have a Wellcome Trust Investigator Award in Science to investigate how this mechanism underpins the role of disordered blood clotting in the pathogenesis of Buruli ulcer.
After graduating from the University of Manchester with a degree in Molecular Biology, I went on to study for a PhD with David Lane at Imperial College London. My early career was in haemostasis (regulation of blood clotting), with a particular focus on the protein C anticoagulant pathway and endothelial cell biology. I had a career break and then joined the Macrophage Biology group at the Kennedy Institute of Rheumatology (Imperial College London), changing my research field to study inflammatory signalling. My first academic appointment here at the University of Surrey started when I recieved my first award from the Wellcome Trust.
Research interests
As a molecular biologist, my research interests concern the molecular detail of the pathogenesis of different disease systems. My group specialises in the uncovering of “unusual” mechanisms of gene regulation in eukaryotic cells; as exemplified by our work studying mycolactone function. In order to understand this fully, we have had to (and to an extent still are) follow a trial of breadcrumbs through the fundamental concepts of molecular biology starting from gene transcription, through protein translation and on to protein fate.
University roles and responsibilities
- Director of Post-Graduate Research for the Department of Microbial Sciences


ResearchResearch interests
The major focus of my research is understanding the mechanism of action of the mycobacterial virulence factor mycolactone. This lipid-like molecule is synthesised by a small group of so-called mycolactone-producing mycobacteria (MPM) which each make subtly different mycolactones. Species in this group include those which are pathogenic to frogs, fish and, importantly, humans. The latter, Mycobacterium ulcerans, causes a disease called Buruli ulcer which is a serious skin infection that affects some of the poorest communities in the world. It causes massive debilitating skin ulcers, often leading to disfigurement or amputation (see http://www.who.int/buruli/en/).
Mycolactone is known to be responsible for the pathogenesis of Buruli ulcer, because it is both immunosuppressive and cytotoxic to host cells. The immunosuppression includes inhibition of the production of cytokines that drive inflammation. We first showed that production of TNF, IL-6 and Cox-2 were supressed by a post-transcriptional mechanism. Detailed investigations then identified the mechanistic target of the molecule: it prevents the translocation of proteins into the endoplasmic reticulum. Most glycosylated and secreted proteins (like cytokines) undergo a process of co-translational translocation via the Sec61 translocon which acts channel separating the two cellular compartments. Because mycolactone blocks this, the proteins get stuck in the cytosol, are recognised as being mislocated, and are destroyed by the proteasome. Therefore, many proteins that would normally pass through the ER, potentially up to 25% of total cellular protein are made as normal in the presence of mycolactone, but are immediately destroyed again. This highly unusual pathogenic molecular mechanism currently seems unique to Buruli ulcer.
We are now exploiting this knowledge in order to develop better treatments for Buruli ulcer, and other diseases where protein translocation plays an important role. We recently showed that endothelial cells are exquisitely sensitive to mycolactone; 7ng/ml mycolactone is sufficient to deplete thrombomodulin from the surface of the cells in under 24hours. Since thrombomodulin is an essential anticoagulant protein, and fibrin is commonly found in the necrotic skin tissue, this has opened up the whole area of disordered haemostasis in Buruli ulcer. I currently hold a Wellcome Trust Investigator Award in Science to fully delve into how mycolactone and changes to blood coagulation drive the pathological process.
Current group members
Belinda Hall (Post-Doc)
Louise Hsieh (Post-Doc)
Jane Newcombe (Post-Doc)
Katherine Corfield (PhD Student)
Jo Butler (Interdisciplinary PhD Student in Medical Illustration)
Aloysius Loglo (Collaborative PhD Student at KCCR, Kumasi, Ghana)
Lucy Eke (Collaborative PhD Student in Virology)
Efi Mavrogiannaki (Collaborative PhD Student in Chemistry)
Elizabeth Gyamfi (Visiting PhD Student from WACCBIP, Accra, Ghana)
Past group members
Joy Ogbechi (PhD Student)
Scott Dos Santos (Euromasters Student)
Sweta Jain (MSc Student)
Research collaborations
Phil Biggin (Oxford): MD Simulations of mycolactone interactions
Adolfo Cavalie (Saarland University Faculty of Medicine): Calcium leak via the Sec61 channel
Mark Field (Dundee): Evolution of the Sec61 translocon in eukaryotes
Matt Higgins (Oxford): Structural Biology
Stephen High (University of Manchester): Protein translocation
Rebecca Hoyle (Southampton: Mathematical modelling of Buruli ulcer lesions
Nick Ktistakis (Babraham): Early stage induction of autophagy by mycolactone
Lydia Mosi (WACCBIP, Ghana): Ultra-long read sequencing of the M. ulcerans genome
Richard Phillips (KCCR, Ghana): Buruli ulcer clinical research
Gerd Pluschke (Swiss Tropical and Public Health Institute): Buruli ulcer pathogenesis
Anne Willis (MRC Toxicology Unit): Translational control
Indicators of esteem
Member of the Wellcome Trust Expert Review Group on Pathogen Biology
Editorial board of Tuberculosis
Member of the Biochemical Society's Awards Committee
Visiting Research Fellow at University of Southampton
Visiting Lecturer at the University of Ghana
The group regularly attends the WHO Global Buruli ulcer Initiative meetings

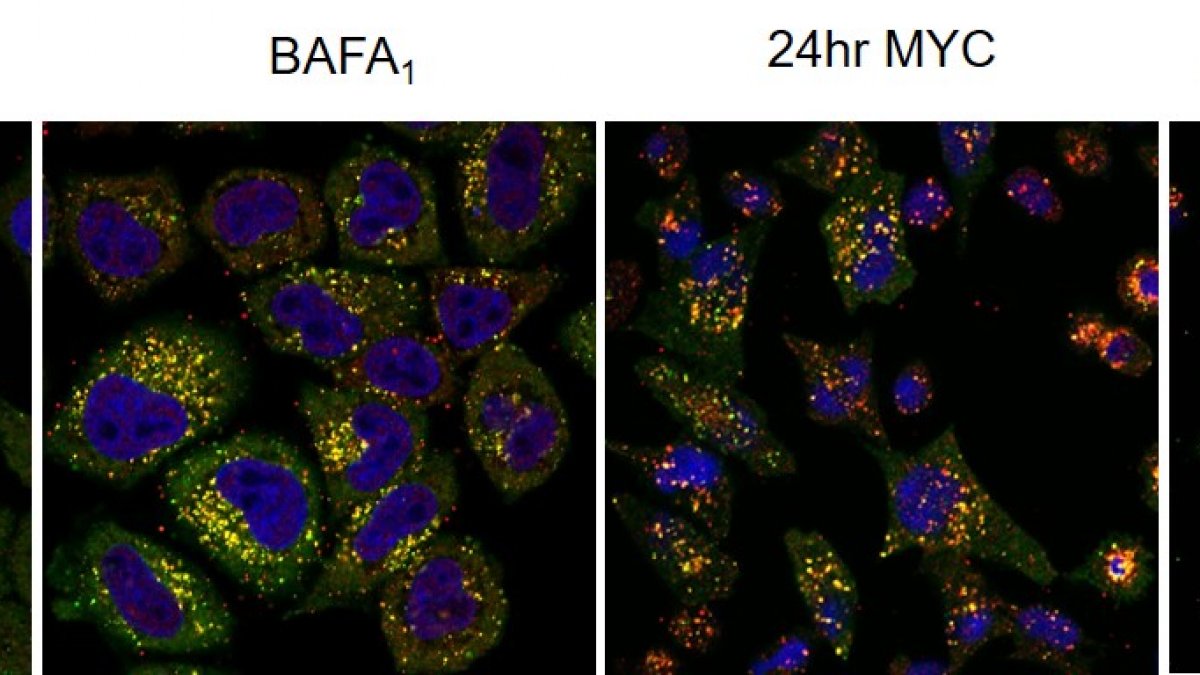
Co-immunofluorescence for p62 (green) and LC3 (red) reveals selective autophagy in mycolactone-exposed cells. The cargo is probably mislocalised/misfolded proteins accumulating in cytosol due to the action of mycoalctone at Sec61
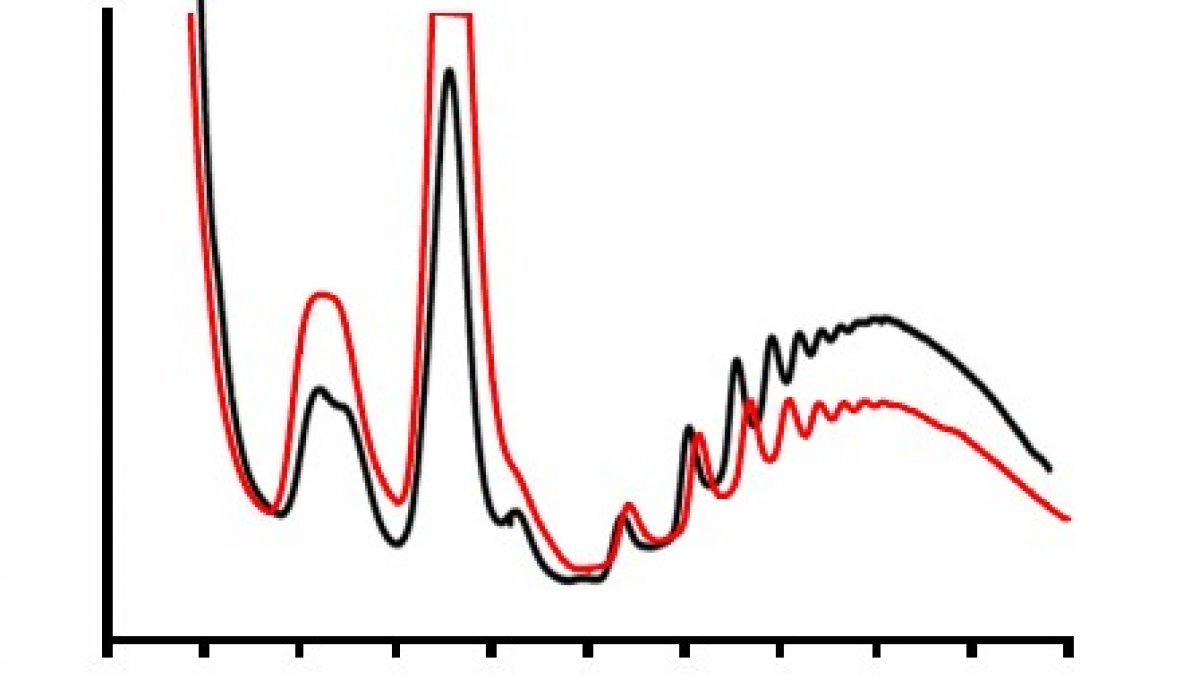
Polysome profiling of normal cells, and those incubated with mycolactone (red) allows us to visualise changes in translation rates of proteins
Research interests
The major focus of my research is understanding the mechanism of action of the mycobacterial virulence factor mycolactone. This lipid-like molecule is synthesised by a small group of so-called mycolactone-producing mycobacteria (MPM) which each make subtly different mycolactones. Species in this group include those which are pathogenic to frogs, fish and, importantly, humans. The latter, Mycobacterium ulcerans, causes a disease called Buruli ulcer which is a serious skin infection that affects some of the poorest communities in the world. It causes massive debilitating skin ulcers, often leading to disfigurement or amputation (see http://www.who.int/buruli/en/).
Mycolactone is known to be responsible for the pathogenesis of Buruli ulcer, because it is both immunosuppressive and cytotoxic to host cells. The immunosuppression includes inhibition of the production of cytokines that drive inflammation. We first showed that production of TNF, IL-6 and Cox-2 were supressed by a post-transcriptional mechanism. Detailed investigations then identified the mechanistic target of the molecule: it prevents the translocation of proteins into the endoplasmic reticulum. Most glycosylated and secreted proteins (like cytokines) undergo a process of co-translational translocation via the Sec61 translocon which acts channel separating the two cellular compartments. Because mycolactone blocks this, the proteins get stuck in the cytosol, are recognised as being mislocated, and are destroyed by the proteasome. Therefore, many proteins that would normally pass through the ER, potentially up to 25% of total cellular protein are made as normal in the presence of mycolactone, but are immediately destroyed again. This highly unusual pathogenic molecular mechanism currently seems unique to Buruli ulcer.
We are now exploiting this knowledge in order to develop better treatments for Buruli ulcer, and other diseases where protein translocation plays an important role. We recently showed that endothelial cells are exquisitely sensitive to mycolactone; 7ng/ml mycolactone is sufficient to deplete thrombomodulin from the surface of the cells in under 24hours. Since thrombomodulin is an essential anticoagulant protein, and fibrin is commonly found in the necrotic skin tissue, this has opened up the whole area of disordered haemostasis in Buruli ulcer. I currently hold a Wellcome Trust Investigator Award in Science to fully delve into how mycolactone and changes to blood coagulation drive the pathological process.
Current group members
Belinda Hall (Post-Doc)
Louise Hsieh (Post-Doc)
Jane Newcombe (Post-Doc)
Katherine Corfield (PhD Student)
Jo Butler (Interdisciplinary PhD Student in Medical Illustration)
Aloysius Loglo (Collaborative PhD Student at KCCR, Kumasi, Ghana)
Lucy Eke (Collaborative PhD Student in Virology)
Efi Mavrogiannaki (Collaborative PhD Student in Chemistry)
Elizabeth Gyamfi (Visiting PhD Student from WACCBIP, Accra, Ghana)
Past group members
Joy Ogbechi (PhD Student)
Scott Dos Santos (Euromasters Student)
Sweta Jain (MSc Student)
Research collaborations
Phil Biggin (Oxford): MD Simulations of mycolactone interactions
Adolfo Cavalie (Saarland University Faculty of Medicine): Calcium leak via the Sec61 channel
Mark Field (Dundee): Evolution of the Sec61 translocon in eukaryotes
Matt Higgins (Oxford): Structural Biology
Stephen High (University of Manchester): Protein translocation
Rebecca Hoyle (Southampton: Mathematical modelling of Buruli ulcer lesions
Nick Ktistakis (Babraham): Early stage induction of autophagy by mycolactone
Lydia Mosi (WACCBIP, Ghana): Ultra-long read sequencing of the M. ulcerans genome
Richard Phillips (KCCR, Ghana): Buruli ulcer clinical research
Gerd Pluschke (Swiss Tropical and Public Health Institute): Buruli ulcer pathogenesis
Anne Willis (MRC Toxicology Unit): Translational control
Indicators of esteem
Member of the Wellcome Trust Expert Review Group on Pathogen Biology
Editorial board of Tuberculosis
Member of the Biochemical Society's Awards Committee
Visiting Research Fellow at University of Southampton
Visiting Lecturer at the University of Ghana
The group regularly attends the WHO Global Buruli ulcer Initiative meetings
Co-immunofluorescence for p62 (green) and LC3 (red) reveals selective autophagy in mycolactone-exposed cells. The cargo is probably mislocalised/misfolded proteins accumulating in cytosol due to the action of mycoalctone at Sec61
Polysome profiling of normal cells, and those incubated with mycolactone (red) allows us to visualise changes in translation rates of proteins
Supervision
Postgraduate research supervision
Aloysius Loglo (2018-current) Collaborative supervisor KCCR, Ghana
The role of diet and coagulation in the pathogenesis of Buruli ulcer
Katherine Corfield (2017-current) Principal supervisor University of Surrey
The evolution of mycolactone-dependent inhibition of the Sec61 translocon
Efi Mavrogiannaki (2017-current) Co-supervisor University of Surrey
New approaches to chemical synthesis of mycolactone
Fatumah Atuhaire (2017-current) Collaborative supervisor University of Southampton
Mathematical modelling of Buruli ulcer lesion formation
Jo Culley (2017-current) Principal supervisor University of Surrey
Utilising medical illustration to improve understanding & communication of disfiguring skin NTDs
Lucy Eke (2017-current) Co-supervisor University of Surrey
Realising the therapeutic potential of exotoxins
Elizabeth Gyamfi (2016-current) Collaborative supervisor University of Ghana
Probing the diversity of Mycolactone-producing mycobacteria using MinION Sequencing
Joy Ogbechi (2013-2016) Principal supervisor University of Surrey
Investigating the mechanisms behind the tissue necrosis in Mycobacterium ulcerans infection
Teaching
Undergraduate
BMS2045: Introduction to Immunology
BMS2036: Molecular Biology and Genetics, from Genes to Biological Function
BMS3054: Clinical Immunology and Immunohaematology
Postgraduate
MSc in Medical Microbiology
MMIM024: Pathogenesis of Infectious Diseases
MMIM027: Research Methods 2 (Module Coordinator)
Publications
Protein S deficiency is a known risk factor for thrombosis. The coexistence of phenotypic type I (reduction in total and free antigen) and type III (reduction in free antigen only) protein S deficiencies in 14 of 18 families was recently reported. We investigated the cause of this phenotypic variation in the largest of these families (122 family members, including 44 affected individuals) using both molecular genetic and phenotypic analysis. We have identified a sole causative mutation (Gly295Val) in three family members representative of the variable phenotype. Complete cosegregation of the mutation with reduced free protein S antigen levels was found, regardless of the total antigen level. Analysis of phenotypic data showed high correlations between total protein S antigen and age in both normal and protein S-deficient family members, irrespective of gender. Free protein S antigen levels were not influenced by age, a finding explained by an association between beta-chain containing C4b-binding protein (C4bBP-beta+) antigen levels and age. We propose that the identified Gly295Val mutation causes quantitative, or type I, protein S deficiency, and that as age increases the total protein S antigen level normalizes with respect to the reference plasma pool, giving rise to a type III protein S-deficient phenotype.
Purpose of the ReviewBuruli ulcer (BU) is a necrotizing and disabling cutaneous disease caused by Mycobacterium ulcerans, one of the skin-related neglected tropical diseases (skin NTDs). This article aims to review the current knowledge of this disease and challenges ahead.Recent FindingsAround 60,000 cases of BU have been reported from over 33 countries between 2002 and 2017. Encouraging findings for development of point-of-care tests for BU are being made, and its treatment is currently in the transition period from rifampicin plus streptomycin (injection) to all-oral regimen. A major recent advance in our understanding of its pathogenesis has been agreement on the mechanism of action of the major virulence toxin mycolactone in host cells, targeting the Sec61 translocon during a major step in protein biogenesis.SummaryBU is distributed mainly in West Africa, but cases are also found in other parts of the world. We may be underestimating its true disease burden, due to the limited awareness of this disease. More awareness and more understanding of BU will surely contribute in enhancing our fight against this skin NTD.
Buruli ulcer is a slowly progressive necrotizing disease of the skin caused by Mycobacterium ulcerans, recognized by WHO as a neglected tropical disease. Around 2,000 cases are reported annually, but underdiagnosis and under-reporting probably obscure the true burden. A major advance in the understanding of Buruli ulcer pathogenesis was the discovery that mycolactone, a lipid-like exotoxin secreted by M. ulcerans, inhibits the Sec61 translocon, driving tissue destruction and immune suppression. M. ulcerans is an opportunistic environmental pathogen; however, its mechanisms of transmission remain unclear in most regions. PCR is the current gold standard for diagnosis, but its cost and technical demands limit use in resource-limited settings. Treatment is available with an oral regimen of rifampicin plus clarithromycin for 8 weeks, but further research is in progress to explore alternative drugs, optimized dosing and duration, and improved affordability. Adjunctive wound care, management of paradoxical reactions and rehabilitation, including physiotherapy and psychosocial support, are essential components of Buruli ulcer management. Future efforts should focus on elucidating transmission pathways to inform prevention, developing rapid diagnostics, refining and adapting drug regimens for diverse clinical presentations and patient groups, and advancing wound care. Strengthening healthcare worker training and integrating Buruli ulcer control with that of other skin diseases will enhance accessibility to early diagnosis and treatment, prevent disabilities and deformities, and reduce stigma, ultimately ensuring better quality of life for affected individuals worldwide.
In 2008, we published the first set of guidelines for standardizing research in autophagy. Since then, this topic has received increasing attention, and many scientists have entered the field. Our knowledge base and relevant new technologies have also been expanding. Thus, it is important to formulate on a regular basis updated guidelines for monitoring autophagy in different organisms. Despite numerous reviews, there continues to be confusion regarding acceptable methods to evaluate autophagy, especially in multicellular eukaryotes. Here, we present a set of guidelines for investigators to select and interpret methods to examine autophagy and related processes, and for reviewers to provide realistic and reasonable critiques of reports that are focused on these processes. These guidelines are not meant to be a dogmatic set of rules, because the appropriateness of any assay largely depends on the question being asked and the system being used. Moreover, no individual assay is perfect for every situation, calling for the use of multiple techniques to properly monitor autophagy in each experimental setting. Finally, several core components of the autophagy machinery have been implicated in distinct autophagic processes (canonical and noncanonical autophagy), implying that genetic approaches to block autophagy should rely on targeting two or more autophagy-related genes that ideally participate in distinct steps of the pathway. Along similar lines, because multiple proteins involved in autophagy also regulate other cellular pathways including apoptosis, not all of them can be used as a specific marker for bona fide autophagic responses. Here, we critically discuss current methods of assessing autophagy and the information they can, or cannot, provide. Our ultimate goal is to encourage intellectual and technical innovation in the field.
Postoperative myopenia (acute muscle loss) is a significant concern following major cancer resection surgery, contributing to prolonged recovery, increased dependency and impaired quality of life. Despite its clinical relevance, the mechanisms underlying postoperative myopenia and its potential mediators remain poorly understood. This study aims to evaluate the acute changes in muscle size, strength and activity following major cancer surgery and to explore the role of insulin resistance and selenium deficiency as potential mediators of these alterations. A prospective cohort study was conducted involving 52 patients undergoing elective open major abdominal surgery for cancer. Preoperative and postoperative assessments included measurements of rectus femoris cross-sectional area (RFCSA) via ultrasound, handgrip strength (HGS), sniff nasal inspiratory pressure (SNIP) and physical activity using an accelerometer. Blood samples were analysed for markers of muscle metabolism, systemic inflammation, insulin resistance and selenium levels. Statistical analyses were performed to compare preoperative and postoperative values and to explore correlations between these measures and clinical outcomes. A significant reduction in RFCSA was observed in 50% of patients, with a median decrease of 10.2% within the first week post-surgery, which persisted at the 6-week follow-up. HGS and SNIP also showed significant declines postoperatively, and reduced physical activity was associated with greater muscle loss. Insulin resistance and postoperative selenium depletion were associated with greater reductions in RFCSA. Major cancer surgery is associated with significant acute muscle loss, which is not fully recovered by 6 weeks postoperatively. Insulin resistance and selenium deficiency may contribute to this muscle loss. Further research is needed to investigate potential interventions to prevent or mitigate postoperative myopenia.
Medical illustrations of: - a Buruli ulcer (BU) nodule on the leg, - a Buruli ulcer (BU) nodule being palpated on the leg, - a comparison of the size of a Buruli ulcer (BU) nodule to a coin, - a Buruli ulcer (BU) at large lesion stage, - a Buruli ulcer (BU) papule on the leg (these are
Medical illustrations of: - an ulcerated primary Yaw, - a Buruli ulcer (BU) nodule on the leg with label, - a Buruli ulcer small lesion located on the calf area of the leg, - a Yaws nodule at primary stage on the leg, - a palpating Buruli ulcer nodule with labels (nodules are palpated to test what the lump feels like, and in the case of BU it feels the same size and firmness as a vegetable pea), - a Buruli ulcer (BU) large lesion on the leg, - a palpating Buruli ucler nodule without labels (nodule has been compared to a pigeon pea grown in Africa to highlight that a BU nodule feels the same size and firmness as a pigeon pea beneath the skin), - a Buruli ulcer small lesion with label, - comparison of a Buruli ulcer (BU) papule with a nodule, - a Buruli ulcer (BU) nodule in cross section within the skin layers (accompanying it is healthy skin layers for comparison), - a Buruli ulcer (BU) at ulcer stage depicting what is in the wound itself, - a Buruli ulcer (BU) plaque with swelling on a single leg and along side it plaque with oedema for comparison, - a Buruli ulcer (BU) and plaque on the leg with no swelling (BU plaque can present in many forms on a patient for example with and without oedema), - a Buruli ulcer (BU) with plaque and oedema on a single leg, - a Buruli ulcer (BU) nodule on the leg. These are all part of a set of skin related neglected topical diseases (skin NTDs), which are chronic infectious diseases found in tropical and sub-tropical regions of the world. How the Illustrations were created. The research of skin neglected tropical diseases (skin NTDs) was combined with the method of medical illustration completing a practice-based and multi-disciplinary PhD study. The resulting medical illustrations include different skin presentations and stages of skin NTDs. The published and clinically approved medical illustrations are being made available to use both in print and digital format. Their purpose to support healthcare professionals for skin NTD identification and detection, and for use within patient education as engaging and informative visual communications for those living in regions such as Sub-Saharan Africa.
Buruli ulcer (BU), caused by Mycobacterium ulcerans, is a devastating necrotizing skin disease. Key to its pathogenesis is mycolactone, the exotoxin virulence factor that is both immunosuppressive and cytotoxic. The discovery that the essential Sec61 translocon is the major cellular target of mycolactone explains much of the disease pathology, including the immune blockade. Sec61 inhibition leads to a loss in production of nearly all cytokines from monocytes, macrophages, dendritic cells and T cells, as well as antigen presentation pathway proteins and costimulatory molecules. However, there has long been evidence that the immune system is not completely incapable of responding to M. ulcerans infection. In particular, IL-1 beta was recently shown to be present in BU lesions, and to be induced from M. ulcerans-exposed macrophages in a mycolactone-dependent manner. This has important implications for our understanding of BU, showing that mycolactone can act as the "second signal " for IL-1 beta production without inhibiting the pathways of unconventional secretion it uses for cellular release. In this Perspective article, we validate and discuss this recent advance, which is entirely in-line with our understanding of mycolactone's inhibition of the Sec61 translocon. However, we also show that the IL-1 receptor, which uses the conventional secretory pathway, is sensitive to mycolactone blockade at Sec61. Hence, a more complete understanding of the mechanisms regulating IL-1 beta function in skin tissue, including the transient intra-macrophage stage of M. ulcerans infection, is urgently needed to uncover the double-edged sword of IL-1 beta in BU pathogenesis, treatment and wound healing.
The drivers of tissue necrosis in Mycobacterium ulcerans infection (Buruli ulcer disease) have historically been ascribed solely to the directly cytotoxic action of the diffusible exotoxin, mycolactone. However, its role in the clinically evident vascular component of disease aetiology remains poorly explained. We have now dissected mycolactone's effects on human primary vascular endothelial cells in vitro. We show that mycolactone-induced changes in endothelial morphology, adhesion, migration, and permeability are dependent on its action at the Sec61 translocon. Unbiased quantitative proteomics identified a profound effect on proteoglycans, driven by rapid loss of type II transmembrane proteins of the Golgi, including enzymes required for glycosaminoglycan (GAG) synthesis, combined with a reduction in the core proteins themselves. Loss of the glycocalyx is likely to be of particular mechanistic importance, since knockdown of galactosyltransferase II (beta-1,3-galactotransferase 6; B3GALT6), the GAG linker-building enzyme, phenocopied the permeability and phenotypic changes induced by mycolactone. Additionally, mycolactone depleted many secreted basement membrane components and microvascular basement membranes were disrupted in vivo during M. ulcerans infection in the mouse model. Remarkably, exogenous addition of laminin-511 reduced endothelial cell rounding, restored cell attachment and reversed the defective migration caused by mycolactone. Hence supplementing mycolactone-depleted extracellular matrix may be a future therapeutic avenue, to improve wound healing rates.
Buruli ulcer (BU) is a skin infection caused by Mycobacterium ulcerans and a neglected tropicaldisease of the skin (skin NTD). Antibiotic treatments are available but, to be effective in theabsence of surgery, BU must be detected at its earliest stages (an innocuous-looking lumpunder the skin) and adherence to prescribed drugs must be high. This study aimed to developmultisensory medical illustrations of BU to support communication with at-risk communities. Weused a Think Aloud method to explore community health workers’ (n¼6) experiences of BUwith a focus on the role of their five senses, since these non-medical disease experts are familiarwith the day-to-day challenges presented by BU. Thematic analysis of the transcripts identifiedthree key themes relating to ‘Detection,’ ‘Help Seeking,’ and ‘Adherence’ with a transcendingtheme ‘Senses as key facilitators of health care’. New medical illustrations, for which we coin thephrase “5D illustrations” (signifying the contribution of the five senses) were then developed toreflect these themes. The senses therefore facilitated an enriched narrative enabling the productionof relevant and useful visuals for health communication. The medical artist communitycould utilise sensory experiences to create dynamic medical illustrations for use in practice.
Background. According to the World Health Organization, neglected tropical diseases (NTDs) affect over two billion people worldwide. While the links between nutrition and many diseases have become clear over recent decades, NTDs have lagged behind and the linkage with nutrition is largely unknown. We conducted this systematic review with meta-analysis to determine the current knowledge on the association between NTDs and malnutrition. Methodology. PubMed, Embase, Scopus and African Journals Online databases were searched using predefined search terms. We included all original articles with a case–control design and at least one NTD. The studies had to compare nutritional parameters between infected cases and control participants. Articles that did not report original data were excluded. The quality of the studies was assessed using the Newcastle–Ottawa scale. Pooled estimates were conducted using the random effect model. The publication bias of the studies was determined by funnel plots. Q and I 2 statistics were used to assess the heterogeneity of the studies. Results. After screening 1294 articles, only 16 qualified for the systematic review and 12 for meta-analysis. These predominately had a focus on soil-transmitted helminthiasis (ascariasis, hookworm diseases and trichuriasis) and schistosomiasis, with a minority concerning leishmaniasis and leprosy. Pooled estimates showed an association between intestinal parasites and stunting in children [odds ratio (OR) = 1.38, 95% confidence interval (CI): 1.14–1.66, I 2 = 0%, tau 2 = 0]. We also identified a moderate association established between serum iron deficiency (OR = 4.67, 95% CI: 1.91–11.44, tau 2 = 0) and intestinal parasites. Conclusions/significance. Of the 20 NTDs, the links between diet and disease have been explored for only 4. There is a paucity of data from low- and middle-income countries and least-developed countries where the NTD burden is high. Therefore, more research into the role of malnutrition in NTDs other than intestinal parasites, leishmaniasis and leprosy is needed.
A medical illustration of Buruli ulcer (BU) at the plaque stage with oedema which is additional swelling. BU is part of a set of skin related neglected topical diseases (skin NTDs), which are chronic infectious diseases found in tropical and sub-tropical regions of the world. The purpose to illustrate how BU plaque with oedema presents on the skin, provided for use within visual communication and for educational purposes regarding skin NTDs. The original purpose for use by health care workers in Ghana, Africa.
A medical illustration of Buruli ulcer (BU) at the small lesion stage. BU is part of a set of skin related neglected topical diseases (skin NTDs), which are chronic infectious diseases found in tropical and sub-tropical regions of the world. The purpose to illustrate how BU at the small lesion stage presents on the skin, provided for use within visual communications and for educational purposes regarding skin NTDs. The original purpose for use by health care workers in Ghana, Africa.
A medical illustration of Buruli ulcer (BU) at the large lesion stage. BU is part of a set of skin related neglected topical diseases (skin NTDs), which are chronic infectious diseases found in tropical and sub-tropical regions of the world. The purpose to illustrate how BU with a large lesion presents on the skin, provided for use within visual communications and for educational purposes regarding skin NTDs. The original purpose for use by health care workers in Ghana, Africa.
Protein secretion in eukaryotes and prokaryotes involves a universally conserved protein translocation chan-nel formed by the Sec61 complex. Unrelated small-molecule natural products and synthetic compoundsinhibit Sec61 with differential effects for different substrates or for Sec61 from different organisms, makingthis a promising target for therapeutic intervention. To understand the mode of inhibition and provide insightinto the molecular mechanism of this dynamic translocon, we determined the structure of mammalian Sec61inhibited by theMycobacterium ulceransexotoxin mycolactone via electron cryo-microscopy. Unexpect-edly, the conformation of inhibited Sec61 is optimal for substrate engagement, with mycolactone wedgingopen the cytosolic side of the lateral gate. The inability of mycolactone-inhibited Sec61 to effectively trans-port substrate proteins implies that signal peptides and transmembrane domains pass through the site occu-pied by mycolactone. This provides a foundation for understanding the molecular mechanism of Sec61 inhib-itors and reveals novel features of translocon function and dynamics.
The innate immune response is a tightly regulated process that reacts rapidly in response to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS). Evidence is accumulating that microRNAs contribute to this, although few studies have examined the early events that constitute the "primary" response. LPS-dependent changes to miRNA expression were studied in primary human monocyte-derived macrophages (1°MDMs). An unbiased screen by microarray was validated by qPCR and a method for the absolute quantitation of miRNAs was also developed, utilising 5' phosphorylated RNA oligonucleotide templates. RNA immunoprecipitation was performed to explore incorporation of miRNAs into the RNA-induced silencing complex (RISC). The effect of miRNA functional inhibition on TNF expression (mRNA and secretion) was investigated. Of the 197 miRNAs expressed in 1°MDMs, only five were induced >1.5-fold. The most strongly induced was miR-155-3p, the partner strand to miR-155-5p, which are both derived from the MIR155HG/BIC gene (pri-miR-155). The abundance of miR-155-3p was induced transiently ~250-fold at 2-4hrs and then returned towards baseline, mirroring pri-miR-155. Other PAMPs, IL-1β, and TNF caused similar responses. IL-10, NF-κB, and JNK inhibition reduced these responses, unlike cytokine-suppressing mycolactone. Absolute quantitation revealed that miRNA abundance varies widely from donor-to-donor, and showed that miR-155-3p abundance is substantially less than miR-155-5p in unstimulated cells. However, at its peak there were 446-1,113 copies/cell, and miR-155-3p was incorporated into the RISC with an efficiency similar to miR-16-5p and miR-155-5p. Inhibition of neither miRNA affected TNF secretion after 2hrs in 1°MDMs, but technical challenges here are noted. Dynamic regulation of miRNAs during the primary response is rare, with the exception of miR-155-3p. Further work is required to establish whether its low abundance, even at the transient peak, is sufficient for biological activity and to determine whether there are specific mechanisms determining its biogenesis from miR-155 precursors.
Ipomoeassin F is a potent natural cytotoxin that inhibits growth of many tumor cell lines with single-digit nanomolar potency. However, its biological and pharmacological properties have remained largely unexplored. Building upon our earlier achievements in total synthesis and medicinal chemistry, we used chemical proteomics to identify Sec61α (protein transport protein Sec61 subunit alpha isoform 1), the pore-forming subunit of the Sec61 protein translocon, as a direct binding partner of ipomoeassin F in living cells. The interaction is specific and strong enough to survive lysis conditions, enabling a biotin analogue of ipomoeassin F to pull down Sec61α from live cells, yet it is also reversible, as judged by several experiments including fluorescent streptavidin staining, delayed competition in affinity pulldown, and inhibition of TNF biogenesis after washout. Sec61α forms the central subunit of the ER protein translocation complex, and the binding of ipomoeassin F results in a substantial, yet selective, inhibition of protein translocation in vitro and a broad ranging inhibition of protein secretion in live cells. Lastly, the unique resistance profile demonstrated by specific amino acid single-point mutations in Sec61α provides compelling evidence that Sec61α is the primary molecular target of ipomoeassin F and strongly suggests that the binding of this natural product to Sec61α is distinctive. Therefore, ipomoeassin F represents the first plant-derived, carbohydrate-based member of a novel structural class that offers new opportunities to explore Sec61α function and to further investigate its potential as a therapeutic target for drug discovery.
Buruli ulcer (BU), one of the skin-related neglected tropical diseases (skin NTDs), is a necrotizing and disabling cutaneous disease caused by subcutaneous infection with Mycobacterium ulcerans. Leading on from the World Health Organization’s (WHO) establishment of a global BU initiative in 1998, >67,000 cases of BU have been reported from over 32 countries, mostly from West Africa and Australia. While treatment is currently in the transition period from rifampicin plus streptomycin (injection) to an all-oral regimen, it cannot hope to eradicate this opportunistic environmental pathogen. M. ulcerans is genetically very similar to related pathogenic organisms M. marinum, M. leprae and M. tuberculosis. However, M. ulcerans carries a unique megaplasmid, pMUM001, encoding the biosynthetic machinery responsible for production of a lipid-like exotoxin virulence factor, mycolactone. This diffusible compound causes the substantial divergence in BU’s pathogenic aetiology from other mycobacterial infections. Hence, mycolactone is cytotoxic and immunosuppressive and causes vascular dysfunction in infected skin. A major recent advance in our understanding of BU pathogenesis has been agreement on the mycolactone’s mechanism of action in host cells, targeting the Sec61 translocon during a major step in secretory and membrane protein biogenesis. While vaccine development for all mycobacteria has been challenging, mycolactone production likely presents a particular challenge in the development of a BU vaccine. The live-attenuated vaccine BCG is known to provide only partial and transient protection in humans but provides a convenient baseline in mouse preclinical studies where it can delay, but not prevent, disease progression. No experimental vaccine strategy has yet conferred greater protection than BCG. However, there is now the prospect of developing a vaccine against mycolactone itself, which may provide hope for the future.
The plant-derived macrocyclic resin glycoside ipomoeassin F (Ipom-F) binds to Sec61 alpha and significantly disrupts multiple aspects of Sec61-mediated protein biogenesis at the endoplasmic reticulum, ultimately leading to cell death. However, extensive assessment of Ipom-F as a molecular tool and a therapeutic lead is hampered by its limited production scale, largely caused by intramolecular assembly of the macrocyclic ring. Here, using in vitro and/or in cellula biological assays to explore the first series of ring-opened analogues for the ipomoeassins, and indeed all resin glycosides, we provide clear evidence that macrocyclic integrity is not required for the cytotoxic inhibition of Sec61-dependent protein translocation by Ipom-F. Furthermore, our modeling suggests that open-chain analogues of Ipom-F can interact with multiple sites on the Sec61 alpha subunit, most likely located at a previously identified binding site for mycolactone and/or the so-called lateral gate. Subsequent in silico-aided design led to the discovery of the stereochemically simplified analogue 3 as a potent, alternative lead compound that could be synthesized much more efficiently than Ipom-F and will accelerate future ipomoeassin research in chemical biology and drug discovery. Our work may also inspire further exploration of ring-opened analogues of other resin glycosides.
Equine viral arteritis is an infectious disease of equids caused by equine arteritis virus (EAV), an RNA virus of the family Arteriviridae. Dendritic cells (DC) are important modulators of the immune response with the ability to present antigen to naïve T cells and can be generated in vitro from monocytes (MoDC). DC are important targets for many viruses and this interaction is crucial for the establishment-or rather not-of an anti-viral immunity. Little is known of the effect EAV has on host immune cells, particularly DC. To study the interaction of eqDC with EAV in vitro, an optimized eqMoDC system was used, which was established in a previous study. MoDC were infected with strains of different genotypes and pathogenicity. Virus replication was determined through titration and qPCR. The effect of the virus on morphology, phenotype and function of cells was assessed using light microscopy, flow cytometry and in vitro assays. This study confirms that EAV replicates in monocytes and MoDC. The replication was most efficient in mature MoDC, but variable between strains. Only the virulent strain caused a significant down-regulation of certain proteins such as CD14 and CD163 on monocytes and of CD83 on mature MoDC. Functional studies conducted after infection showed that EAV inhibited the endocytic and phagocytic capacity of Mo and mature MoDC with minimal effect on immature MoDC. Infected MoDC showed a reduced ability to stimulate T cells. Ultimately, EAV replication resulted in an apoptosis-mediated cell death. Thus, EAV evades the host anti-viral immunity both by inhibition of antigen presentation early after infection and through killing infected DC during replication.
Mycolactone is the exotoxin virulence factor of Mycobacterium ulcerans that causes the neglected tropical disease Buruli ulcer. We recently showed it to be a broad spectrum inhibitor of Sec61-dependent co-translational translocation of proteins into the endoplasmic reticulum (ER). An outstanding question is the molecular pathway linking this to its known cytotoxicity. We have now used translational profiling to better understand the reprogramming that occurs in cells exposed to mycolactone. Gene ontology identified enrichment in genes involved in cellular response to stress, and apoptosis signalling amongst those showing enhanced translation. Validation of these results supports a mechanism by which mycolactone activates an integrated stress response meditated by phosphorylation of eIF2α via multiple kinases (PERK, GCN, PKR) without activation of the ER stress sensors IRE1 or ATF6. The response therefore uncouples the integrated stress response from ER stress, and features translational and transcription modes of genes expression that feature the key regulatory transcription factor ATF4. Emphasizing the importance of this uncoupled response in cytotoxicity, downstream activation of this pathway is abolished in cells expressing mycolactone-resistant Sec61α variants. Using multiple genetic and biochemical approaches, we demonstrate that eIF2α phosphorylation is responsible for mycolactone-dependent translation attenuation, which initially protects cells from cell death. However, chronic activation without stress remediation enhances autophagy and apoptosis of cells by a pathway facilitated by ATF4 and CHOP. Our findings demonstrate that priming events at the ER can result in the sensing of stress within different cellular compartments.
Mycolactone is the exotoxin virulence factor produced by Mycobacterium ulcerans, the pathogen responsible for Buruli ulcer. The skin lesions and immunosuppression characteristic of this disease result from the action of mycolactone, which targets the Sec61 complex and inhibits the co-translational translocation of secretory proteins into the endoplasmic reticulum. In this study, we investigate the effect of mycolactone on the Sec61-dependent biogenesis of different classes of transmembrane protein (TMP). Our data suggest that the effect of mycolactone on TMP biogenesis depends on how the nascent chain initially engages the Sec61 complex. For example, translocation of TMP lumenal domains driven by an N-terminal, cleavable signal sequence is efficiently inhibited by mycolactone. In contrast, the effect of mycolactone on protein translocation driven solely by a non-cleavable signal anchor/transmembrane domain depends on which flanking region is translocated. For example, while translocation of the region N-terminal to a signal anchor/transmembrane domain is refractive to mycolactone, C-terminal translocation is efficiently inhibited. Our findings highlight the diversity of Sec61-dependent translocation and provide a molecular basis for understanding the effect of mycolactone on the biogenesis of different TMPs.
Nonmycobacterial (NTM) lung infections are emerging as global health threats; however, the molecular mechanisms underlying host-microbial interactions are poorly understood compared to tuberculosis caused by Mycobacterium tuberculosis. NTM-host interactions are complex and dynamic processes between the mycobacterial components and host factors, influencing the infection outcomes. While NTM evolves numerous strategies to establish the infection and evade from host’s defense system, the host cells encounter the pathogenic stresses through a range of typical (immune) and non-typical components to induce defensive pathways that limit or eradicate intracellular pathogenic replications. Here, a special issue of articles discusses how NTM bacteria modulate the host defense system, in which immune and nonimmune components are functionally involved in the antimicrobial responses, and how different host cell types participate in the protective responses against NTM infections. In doing so, we outline several weapons harboring the therapeutic potential in a tug-of-war at the interface of host and NTM bacteria. A more detailed understanding of underlying NTM-host crosstalks will provide new alternative therapeutic and preventive strategies for NTM infections, which are often refractory to the conventional antibiotic-based regiments (1).
Buruli Ulcer (BU) is the third most common mycobacterium disease following only tuberculosis and leprosy. Though BU is thought to be associated with large-and small-scale disturbances to the landscape and bodies of water frequented by human populations, primary prevention of BU is difficult because the mode of transmission is not known. This chapter reviews the most common environmental risk factors for BU and recent research into understanding its transmission. It is predicted that the proteins affected by mycolactone may share an underlying mechanism of production that could explain their co-regulation. Early work on the mechanism of suppression by mycolac-tone was carried out in Jurkat T cells using ASLs and focused on the suppression of IL-2 production. A multidisciplinary approach to treatment and patient care is essential for optimizing treatment outcomes. Physiotherapy is paramount minimizing and/or preventing disabilities.
Background: The innate immune response is a tightly regulated process that reacts rapidly in response to pathogen-associated molecular patterns(PAMPs) such as lipopolysaccharide (LPS). Evidence is accumulating thatmicroRNAs contribute to this, although few studies have examined the earlyevents that constitute the “primary” response. LPS-dependent changes to miRNA expression were studied inMethods:primary human monocyte-derived macrophages (1°MDMs). An unbiasedscreen by microarray was validated by qPCR and a method for the absolutequantitation of miRNAs was also developed, utilising 5’ phosphorylatedRNA oligonucleotide templates. RNA immunoprecipitation was performedto explore incorporation of miRNAs into the RNA-induced silencingcomplex (RISC). The effect of miRNA functional inhibition on TNFexpression (mRNA and secretion) was investigated. Of the 197 miRNAs expressed in 1°MDMs, only five were inducedResults:>1.5-fold. The most strongly induced was miR-155-3p, the partner strand tomiR-155-5p, which are both derived from the MIR155HG/BIC gene(pri-miR-155). The abundance of miR-155-3p was induced transiently~250-fold at 2-4hrs and then returned towards baseline, mirroringpri-miR-155. Other PAMPs, IL-1β, and TNF caused similar responses.IL-10, NF-κB, and JNK inhibition reduced these responses,unlike cytokine-suppressing mycolactone. Absolute quantitation revealedthat miRNA abundance varies widely from donor-to-donor, and showed thatmiR-155-3p abundance is substantially less than miR-155-5p inunstimulated cells. However, at its peak there were 446-1,113 copies/cell,and miR-155-3p was incorporated into the RISC with an efficiency similar tomiR-16-5p and miR-155-5p. Inhibition of neither miRNA affected TNFsecretion after 2hrs in 1°MDMs, but technical challenges here are noted. Dynamic regulation of miRNAs during the primary responseConclusions:is rare, with the exception of miR-155-3p. Further work is required toestablish whether its low abundance, even at the transient peak, issufficient for biological activity and to determine whether there are specificmechanisms determining its biogenesis from miR-155 precursor.
Ipomoeassin F is a potent natural cytotoxin that inhibits growth of many tumor cell lines with single-digit nanomolar potency. However, its biological and pharmacological properties have remained largely unexplored. Building upon our earlier achievements in total synthesis and medicinal chemistry, we used chemical proteomics to identify Sec61α (protein transport protein Sec61 subunit alpha isoform 1), the pore-forming subunit of the Sec61 protein translocon, as a direct binding partner of ipomoeassin F in living cells. The interaction is specific and strong enough to survive lysis conditions, enabling a biotin analogue of ipomoeassin F to pull down Sec61α from live cells, yet it is also reversible, as judged by several experiments including fluorescent streptavidin staining, delayed competition in affinity pulldown, and inhibition of TNF biogenesis after washout. Sec61α forms the central subunit of the ER protein translocation complex, and the binding of ipomoeassin F results in a substantial, yet selective, inhibition of protein translocation in vitro and a broad ranging inhibition of protein secretion in live cells. Lastly, the unique resistance profile demonstrated by specific amino acid single-point mutations in Sec61α provides compelling evidence that Sec61α is the primary molecular target of ipomoeassin F and strongly suggests that the binding of this natural product to Sec61α is distinctive. Therefore, ipomoeassin F represents the first plant-derived, carbohydrate-based member of a novel structural class that offers new opportunities to explore Sec61α function and to further investigate its potential as a therapeutic target for drug discovery.
Buruli ulcer (BU) is a neglected tropical disease caused by subcutaneous infection with Mycobacterium ulcerans and its exotoxin mycolactone. BU displays coagulative necrosis and widespread fibrin deposition in affected skin tissues. Despite this, the role of the vasculature in BU pathogenesis remains almost completely unexplored. We hypothesise that fibrin-driven ischemia can be an 'indirect' route to mycolactone-dependent tissue necrosis by a mechanism involving vascular dysfunction. Here, we tracked >900 vessels within contiguous tissue sections from eight BU patient biopsies. Our aim was to evaluate their vascular and coagulation biomarker phenotype and explore potential links to fibrin deposition. We also integrated this with our understanding of mycolactone's mechanism of action at Sec61 and its impact on proteins involved in maintaining normal vascular function. Our findings showed that endothelial cell dysfunction is common in skin tissue adjacent to necrotic regions. There was little evidence of primary haemostasis, perhaps due to mycolactone-dependent depletion of endothelial von Willebrand factor. Instead, fibrin staining appeared to be linked to the extrinsic pathway activator, tissue factor (TF). There was significantly greater than expected fibrin staining around vessels that had TF staining within the stroma, and this correlated with the distance it extended from the vessel basement membrane. TF-induced fibrin deposition in these locations would require plasma proteins outside of vessels, therefore we investigated whether mycolactone could increase vascular permeability in vitro. This was indeed the case, and leakage was further exacerbated by IL-1β. Mycolactone caused the loss of endothelial adherens and tight junctions by the depletion of VE-cadherin, TIE-1, TIE-2 and JAM-C; all Sec61-dependent proteins. Taken together, our findings suggest that both vascular and lymphatic vessels in BU lesions become "leaky" during infection, due to the unique action of mycolactone, allowing TF-containing structures and plasma proteins into skin tissue, ultimately leading to local coagulopathy and tissue ischemia.
Mycolactone is a polyketide macrolide lipid-like secondary metabolite synthesized by Mycobacterium ulcerans, the causative agent of BU (Buruli ulcer), and is the only virulence factor for this pathogen identified to date. Prolonged exposure to high concentrations of mycolactone is cytotoxic to diverse mammalian cells (albeit with varying efficiency), whereas at lower doses it has a spectrum of immunosuppressive activities. Combined, these pleiotropic properties have a powerful influence on local and systemic cellular function that should explain the pathophysiology of BU disease. The last decade has seen significant advances in our understanding of the molecular mechanisms underlying these effects in a range of different cell types. The present review focuses on the current state of our knowledge of mycolactone function, and its molecular and cellular targets, and seeks to identify commonalities between the different functional and cellular systems. Since mycolactone influences fundamental cellular processes (cell division, cell death and inflammation), getting to the root of how mycolactone achieves this could have a profound impact on our understanding of eukaryotic cell biology.
Toll-like receptors (TLRs) are central to innate immunity and yet their expression is widespread and not restricted to professional inflammatory cells. TLRs have been reported on adipocytes and have been implicated in obesity-associated pathologies such as diabetes. Why TLRs are found on adipocytes is not clear although one hypothesis is that they may coordinate energy utilization for the energy intensive process of an immune response. We have explored TLR signalling in primary human in vitro differentiated adipocytes and investigated the specific adapter molecules that are involved. Only lipopolysaccharide (LPS), poly(I:C), Pam3CSK4 and MALP-2 could induce the production of IL-6, IL-8 and MCP-1 by adipocytes. Poly(I:C) alone caused a strong induction of type I interferons, as assessed by IP-10 production. Using siRNA, it was confirmed that LPS-dependent signalling in adipocytes occurs via TLR4 utilizing the adapter molecules MyD88, Mal and TRIF and caused rapid degradation of IκBα. Pam3CSK4 signalling utilized TLR2, MyD88 and Mal (but not TRIF). However, the response to poly(I:C) observed in these cells appeared not to require TRIF, but MyD88 was required for induction of NFκB-dependent cytokines by Poly(I:C). Despite this, IκBα degradation could not be detected in poly(I:C) stimulated adipocytes at any time-point up to 4 h. Indeed, IL-6 transcription was not induced until 8-16 h after exposure. These data suggest that Pam3CSK4 and LPS signal via the expected routes in human adipocytes, whereas poly(I:C)/TLR3 signalling may act via a TRIF-independent, MyD88-dependent route.
The drivers of tissue necrosis in Mycobacterium ulcerans infection (Buruli ulcer disease) have historically been ascribed solely to the directly cytotoxic action of the diffusible exotoxin, mycolactone. However, its role in the clinically-evident vascular component of disease aetiology remains poorly explained. We have now dissected mycolactone's effects on primary vascular endothelial cells in vitro and in vivo. We show that mycolactone-induced changes in endothelial morphology, adhesion, migration, and permeability are dependent on its action at the Sec61 translocon. Unbiased quantitative proteomics identified a profound effect on proteoglycans, driven by rapid loss of type II transmembrane proteins of the Golgi, including enzymes required for glycosaminoglycan (GAG) synthesis, combined with a reduction in the core proteins themselves. Loss of the glycocalyx is likely to be of particular mechanistic importance, since knockdown of galactosyltransferase II (beta-1,3-galactotransferase 6; B3Galt6), the GAG linker-building enzyme, phenocopied the permeability and phenotypic changes induced by mycolactone. Additionally, mycolactone depleted many secreted basement membrane components and microvascular basement membranes were disrupted in vivo. Remarkably, exogenous addition of laminin-511 reduced endothelial cell rounding, restored cell attachment and reversed the defective migration caused by mycolactone. Hence supplementing mycolactone-depleted extracellular matrix may be a future therapeutic avenue, to improve wound healing rates. Competing Interest Statement The authors have declared no competing interest.
A well-known histopathological feature of diseased skin in Buruli ulcer (BU) is coagulative necrosis caused by the Mycobacterium ulcerans macrolide exotoxin mycolactone. Since the underlying mechanism is not known, we have investigated the effect of mycolactone on endothelial cells, focussing on the expression of surface anticoagulant molecules involved in the protein C anticoagulant pathway. Congenital deficiencies in this natural anticoagulant pathway are known to induce thrombotic complications such as purpura fulimans and spontaneous necrosis. Mycolactone profoundly decreased thrombomodulin (TM) expression on the surface of human dermal microvascular endothelial cells (HDMVEC) at doses as low as 2ng/ml and as early as 8hrs after exposure. TM activates protein C by altering thrombin's substrate specificity, and exposure of HDMVEC to mycolactone for 24 hours resulted in an almost complete loss of the cells' ability to produce activated protein C. Loss of TM was shown to be due to a previously described mechanism involving mycolactone-dependent blockade of Sec61 translocation that results in proteasome-dependent degradation of newly synthesised ER-transiting proteins. Indeed, depletion from cells determined by live-cell imaging of cells stably expressing a recombinant TM-GFP fusion protein occurred at the known turnover rate. In order to determine the relevance of these findings to BU disease, immunohistochemistry of punch biopsies from 40 BU lesions (31 ulcers, nine plaques) was performed. TM abundance was profoundly reduced in the subcutis of 78% of biopsies. Furthermore, it was confirmed that fibrin deposition is a common feature of BU lesions, particularly in the necrotic areas. These findings indicate that there is decreased ability to control thrombin generation in BU skin. Mycolactone's effects on normal endothelial cell function, including its ability to activate the protein C anticoagulant pathway are strongly associated with this. Fibrin-driven tissue ischemia could contribute to the development of the tissue necrosis seen in BU lesions.
The virulence factor mycolactone is responsible for the immunosuppression and tissue necrosis that characterise Buruli ulcer, a disease caused by infection with Mycobacterium ulcerans. In this study, we confirm that Sec61, the protein-conducting channel that coordinates entry of secretory proteins into the endoplasmic reticulum, is a primary target of mycolactone, and characterise the nature of its inhibitory effect. We conclude that mycolactone constrains the ribosome-nascent chain-Sec61 complex, consistent with its broad-ranging perturbation of the co-translational translocation of classical secretory proteins. In contrast, the effect of mycolactone on the post-translational, ribosome-independent translocation of short secretory proteins through the Sec61 complex is dependent on both signal sequence hydrophobicity and the translocation competence of the mature domain. Changes to protease sensitivity strongly suggest that mycolactone acts by inducing a conformational change in the pore-forming Sec61α subunit. These findings establish that mycolactone inhibits Sec61-mediated protein translocation and highlight differences between the co- and post-translational routes that the Sec61 complex mediates. We propose that mycolactone also provides a useful tool for further delineating the molecular mechanisms of Sec61-dependent protein translocation.
The Mycobacterium ulcerans exotoxin, mycolactone, is responsible for the immunosuppression and tissue necrosis that characterizes Buruli ulcer. Mycolactone inhibits SEC61-dependent co-translational translocation of proteins into the endoplasmic reticulum and the resultant cytosolic translation triggers degradation of mislocalized proteins by the ubiquitin-proteasome system. Inhibition of SEC61 by mycolactone also activates multiple EIF2S1/eIF2α kinases in the integrated stress response (ISR). Here we show mycolactone increased canonical markers of selective macroautophagy/autophagy LC3B-II, ubiquitin and SQSTM1/p62 in diverse disease-relevant primary cells and cell lines. Increased formation of puncta positive for the early autophagy markers WIPI2, RB1CC1/FIP200 and ATG16L1 indicates increased initiation of autophagy. The mycolactone response was SEC61A1-dependent and involved a pathway that required RB1CC1 but not ULK. Deletion of Sqstm1 reduced cell survival in the presence of mycolactone, suggesting this response protects against the increased cytosolic protein burden caused by the toxin. However, reconstitution of baseline SQSTM1 expression in cells lacking all autophagy receptor proteins could not rescue viability. Translational regulation by EIF2S1 in the ISR plays a key role in the autophagic response to mycolactone. Mycolactone-dependent induction of SQSTM1 was reduced in eif2ak3−/-/perk−/- cells while the p-EIF2S1 antagonist ISRIB reversed the upregulation of SQSTM1 and reduced RB1CC1, WIPI2 and LC3B puncta formation. Increased SQSTM1 staining could be seen in Buruli ulcer patient skin biopsy samples, reinforcing genetic data that suggests autophagy is relevant to disease pathology. Since selective autophagy and the ISR are both implicated in neurodegeneration, cancer and inflammation, the pathway uncovered here may have a broad relevance to human disease.
The Mycobacterium ulcerans exotoxin, mycolactone, is an inhibitor of co-translational translocation via the Sec61 complex. Mycolactone has previously been shown to bind to, and alter the structure of, the major translocon subunit Sec61α, and change its interaction with ribosome nascent chain complexes. In addition to its function in protein translocation into the ER, Sec61 also plays a key role in cellular Ca2+ homeostasis, acting as a leak channel between the endoplasmic reticulum (ER) and cytosol. Here, we have analysed the effect of mycolactone on cytosolic and ER Ca2+ levels using compartment-specific sensors. We also used molecular docking analysis to explore potential interaction sites for mycolactone on translocons in various states. These results show that mycolactone enhances the leak of Ca2+ ions via the Sec61 translocon, resulting in a slow but substantial depletion of ER Ca2+. This leak was dependent on mycolactone binding to Sec61α because resistance mutations in this protein completely ablated the increase. Molecular docking supports the existence of a mycolactone-binding transient inhibited state preceding translocation and suggests mycolactone may also bind Sec61α in its idle state. We propose that delayed ribosomal release after translation termination and/or translocon “breathing” during rapid transitions between the idle and intermediate-inhibited states allow for transient Ca2+ leak, and mycolactone’s stabilisation of the latter underpins the phenotype observed.
Additional publications
Additional key publications
Simmonds RE and Foxwell BM. NF-kB and its relevance to arthritis and inflammation (Review). Rheumatology (Oxford) 2008: 47; 584-90
Rance J, Follows GA, Cockerill PN, Bonifer C, Lane DA, Simmonds RE. Regulation of the human endothelial cell protein C receptor gene promoter by multiple Sp1 binding sites. Blood 2003:101; 4393-4401
Rezende SM, Lane DA, Mille-Baker B, Samama M, Conard J and Simmonds RE. Protein S Gla-domain mutations causing impaired Ca2+-induced phospholipid binding and severe functional protein S deficiency. Blood 2002: 100; 2812-9
Simmonds RE and Lane DA. The Endothelial Cell Protein C Receptor: A Candidate Genetic Risk Factor for Thrombosis (Invited Commentary). Thromb Haemost 2001: 86; 939-41
Simmonds RE and Lane DA. Structural and functional implications of the intron/exon organisation of the human endothelial cell protein C/activated protein C receptor (EPCR) gene. Comparison with the structure of CD1/Major Histocompatibility Complex alpha1 and alpha2 domains. Blood 1999: 94; 632-41
Simmonds RE, Ireland H, Lane DA, Zöller B, García de Frutos P and Dahlbäck B . Clarification of the risk of venous thrombosis associated with hereditary protein S deficiency by investigation of a large kindred with a characterised gene defect. Ann Int Med 1998; 128; 8-14
Simmonds RE, Zöller B, Ireland H, Thompson E, García de Frutos P, Dahlbäck B and Lane DA. Genetic and phenotypic analysis of a large (122 member) protein S-deficient kindred provides an explanation for the familial coexistence of type I and type III plasma phenotypes. Blood 1997; 89; 4364-70